Why pluripotent stem cells?
Pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), can be guided and differentiated into cells of all lineages to generate every building block of tissues and organs. It is therefore possible to model human or animal development and disease progression in a petri dish to identify novel therapeutic strategies. Importantly, the therapy can be tailored to the patient’s specific disease or mutational context. This type of personalized regenerative medicine can potentially lead to the treatment of a variety of disorders.
PSCs can be guided to follow developmental logics in a dish
Following fertilization, the early mammalian embryonic development process constitutes a rapid series of well-coordinated cellular events that are essential to set the organism’s body plan. During this process, the temporal and spatial coordination between multiple cell types and tissues is particularly important because the variation in the relative timing of these processes can have serious consequences for the health and well-being of an organism.
We are interested in a fascinating feature about development—the developmental timing among individuals of the same species is highly reproducible. Between different species, however, developmental timing varies drastically; i.e., human gestation is roughly 40 weeks, while mice require only 3 weeks. What is the genetic basis that regulates species-specific developmental timing is largely unknown and remains an enigma.
In vitro models that faithfully recapitulate species-specific developmental clocks will be critical to understand the molecular mechanisms that regulates developmental timing. Remarkably, the differentiation rate of species-specific PSCs, largely recapitulates species-specific developmental timing. Thus, many differentiation protocols using human or non-human primate PSCs may take several months or sometimes years to complete, reflecting a longer gestation period. On the other hand, murine PSCs complete the same events in vitro in just days or weeks, reflecting a three-week gestation period. These observations suggest that without normal developmental cues from an intact embryonic or extraembryonic component, a largely unknown intrinsic timing mechanism exists in PSCs in vitro. Our long-term research interest is to understand the developmental timing mechanisms that control cell fate during differentiation and embryonic development.
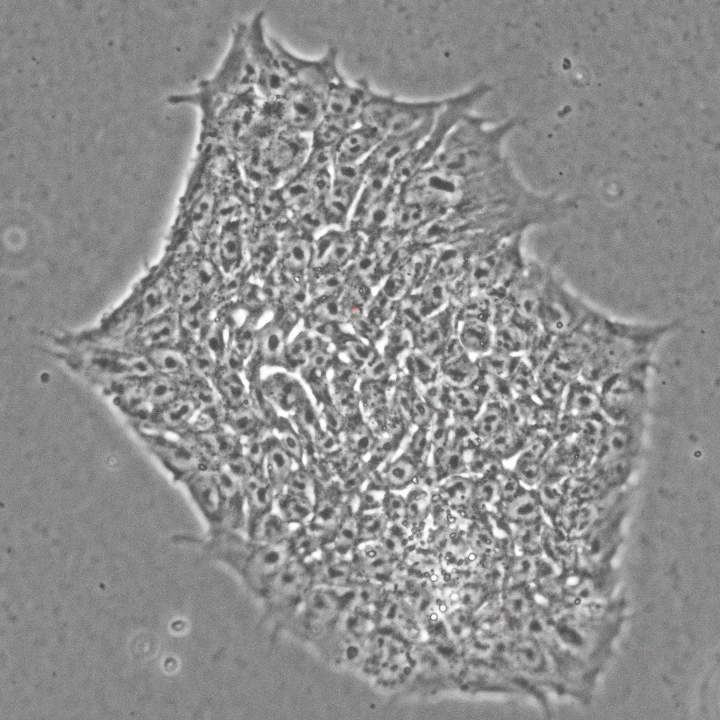
An example of spontaneous beating cradiomyocytes derived from human ESCs in a dish:
Research Projects
In vitro segmentation clock model — a window to understand developmental timing

To identify the principles governing temporal developmental program, we are currently focused on the dynamic function of the segmentation clock, which controls somitogenesis with a species-specific periodicity. Recently, we established in vitro segmentation clock models using human ESC-derived PSM cells, displaying a human-specific gene oscillatory periodicity recapitulating segmentation clock in vivo. Equipped with these novel systems, we are now poised to make significant insights and ask key questions such as: What controls the tempo of a developmental clock? What are the extrinsic or intrinsic factors that could influence developmental timing? How do cells translate this temporal code into spatial identity information during development?
Modeling congenital spinal deformity in a dish — a model to search for novel therapeutic solutions.

A. An illustration of a Carnegie stage 10 human embryo (adapted from Grey H., 1918). B. An X-Ray radiograph of an affected individual with SCDO4 (adapted from Sparrow D. et al., 2008). Insert shows the normal morphology of the spine and rib.
Defects in segmentation result in congenital vertebral anomalies, which are estimated to affect approximately 1 in 1000 live births. Those conditions include: 1) spondylocostal dysostosis (SCDO), a severe but rare condition with misshaped or fused vertebrae, and 2) congenital scoliosis (CS), a prevalent but less severe condition with lateral curvature of the spine. In both conditions, mutations have been identified primarily in the conserved NOTCH signaling pathway. However, due to the lack of proper human cell models, little is known about how these mutations or any additional environmental factors (e.g., temperature, hypoxia) disrupt the process of segmentation. To meet this need, we used genome editing to establish an in vitro model of SCDO4 in which human ESCs harbor the exact missense point mutation in exon 2 of HES7 (OMIM:608059), resulting in an Arg-to-Trp (R25W) substitution at an evolutionarily conserved residue in the DNA-binding domain (HES7R25W). We will use our models to: 1) serve as a cell-based platform for screening novel regulators to restore normal gene oscillation and 2) effectively build additional SCDO/CS models to investigate the influence of environmental factors on the penetrance of CS etiology.
Species-specific reprogramming — an opportunity to compare species-specific pluripotency and functional derivatives.

We are interested in developing iPSCs from mammalian species with various gestation length for comparative developmental biology. Because the signaling requirement for maintaining the pluripotent state could be species-specific, standard mouse or human PSC protocols may not support other species directly. This presents both challenges and opportunities to explore the evolutionary conserved and unique features of pluripotency and cellular reprogramming.